
概述
先天性巨结肠为一错误的命名,因为巨结肠改变不是先天性的。由于巨结肠的远端肠壁内没有神经节细胞,处于痉挛狭窄状态,丧失蠕动和排便功能,致使近端结肠蓄便、积气,而续发扩张、肥厚,逐渐形成了巨结肠改变。 1886年丹麦医生Harald Hirschsprung报道7个月和11个月2例病儿,详细描述了便秘症状和死后扩张结肠的肉眼所见,2年后该文章发表,所以也将该症称为赫什朋病(HirschSprung disease,HD)。由于他认为病变部位在巨结肠,故先天性巨结肠这一病名沿用至今。目前有些文献已将该病称为无神经节细胞症(aganglionosis),或无神经节细胞性巨结肠(agangli...[详细]
病因
1.胚胎学 Bodian认为,先天性巨结肠症的肠壁内神经节细胞缺如是一种壁内神经发育停顿,致使外胚层神经纤维无法参与正常的壁内神经丛发育。1954年Yntema和Hamman在胚胎研究发现,消化道的内在神经丛是由中枢神经嵴衍生而来。其神经母细胞沿已发育的迷走神经干迁移至整个消化道壁内,由头端之食管直至尾端之直肠,此即单相发育学说。而Tam等则提出神经节细胞系由口和肛门向中心发育,此即双相发育学说。 1967年Okamoto等对18例胚胎和胎儿进行了研究,发现肌间神经丛系由神经嵴的神经母细胞形成。这些神经母细胞于胚胎第5周开始沿迷走神经干由头侧向尾侧迁移,于第12周达到消化道远端。在...[详细]
发病机制
1.病理及神经免疫组化改变 先天性巨结肠症的受累肠段可以见到典型的改变,即明显的狭窄段和扩张段。狭窄段位于扩张段远端,一般位于直肠乙状结肠交界处以远,距肛门7~10cm以内。狭窄肠管细小,与扩大肠管直径相差悬殊,其表面结构无甚差异。在与扩大结肠连接部形成漏斗状的移行区(即扩张段远端移行区),此区原属狭窄段,由于近端肠管的蠕动,推挤肠内容物向前移动,长期的挤压促使狭窄段近端肠管扩大成漏斗形。扩张段多位于乙状结肠,严重者可波及横结肠。该肠管异常扩大,其直径较正常增大2~3倍,最大者可达10cm以上。肠壁肥厚、质地坚韧如皮革状。肠管表面失去红润光泽,略呈苍白。结肠带变宽而肌纹呈纵行条状被分裂。结肠...[详细]
临床表现
1.新生儿和婴幼儿 正常新生儿几乎全部在生后24h内排出第一次胎便。患儿则表现为生后不排胎便,开始排胎便及排空时间均推迟。生后2~3天内出现部分甚至完全性肠梗阻症状。患儿呕吐,吐物含胆汁或粪便样液体,并有腹胀及便秘。肛门指诊或用温盐水洗肠,可排出大量胎便及气体,症状缓解。缓解数天后,腹胀、便秘又复出现,又需洗肠才能排便。体检可完全正常,有时可扪到扩张的肠管横贯于上腹部。如果看到有从右向左的蠕动波,可以推测是在无神经节细胞肠段近端扩张的横结肠。腹胀缓解后可以进食,惟多数患儿由于呕吐、拒食而体重不增,发育较差。 在新生儿,也可能以腹泻为突出症状而伴有肠梗阻,经常便秘与腹泻交替出现。如反复...[详细]
并发症
小肠结肠炎及肠穿孔是先天性巨结肠的常见并发症,亦是引起死亡最多见的原因。有文献统计有20%~50%的病儿并发小肠结肠炎,其病死率约30%。肠炎可以发生在各种年龄,但以3个月以内婴儿发病率最高。90%的肠炎病例发生于2岁以内,以后逐渐减少。即使在根治术后或结肠造瘘术后亦偶有出现结肠炎者。Shono报道术后发生肠炎者占61%而多见于Boley手术后。Soaue报道Sweorson术后肠炎占11.6%。Ikeda报道术后肠炎占33.7%,Soalce术后为19.5%,Boley术后12.1%。因此术后预防治疗肠炎成为重要课题。有作者统计先行造瘘术而后发生肠炎者,病死率可以降低。引起肠炎的原因和机制至...[详细]
实验室检查
1.直肠肌层活检 Swenson(1955)最先采用该法,准确率为98%。从直肠壁取肌层活检,证实肌间神经节细胞缺如诊断先天性巨结肠,理论上是最可靠的方法,但因存在一些缺点,故目前并非必要:①Smith(1968)经组织学检查发现,生后神经节细胞有一发育和成熟过程,直肠肌间丛尤其明显,黏膜下丛又落后几周,如不注意,可把正常儿诊断为先天性巨结肠或同源病。②正常直肠在齿状线上方有一低神经节细胞区,在该区内取材可把正常儿诊断为先天性巨结肠或同源病。故强调取材高度在齿状线上方至少新生儿2cm,1岁以内2.5cm,1~3岁3cm,4岁以上3.5cm,如此,短段型巨结肠病儿又易漏诊。③小儿肌层活检有肠穿...[详细]
其他辅助检查
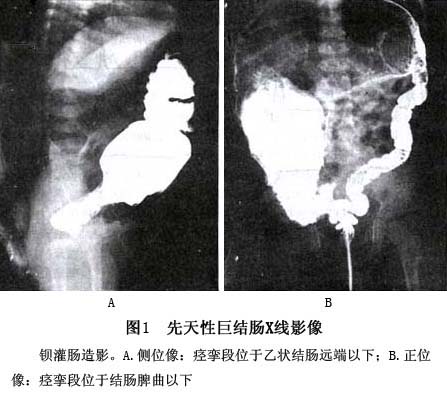
1. X线钡剂灌肠 X线下钡剂灌肠是判定病变范围和选择术式的重要依据。钡剂灌肠目的是显示痉挛段及其上方的扩张段,因此确认扩张段即可,不要过多灌入钡剂继续向上检查,以免加重病儿腹胀及其危险。 痉挛段范围在降结肠以下者,侧位显示最清,故一般仅摄带肛门标记的侧位X线片,但痉挛段达乙状结肠时从正位观察才能全面(图1)。X线钡剂灌肠的诊断率,目前仍徘徊在90%左右,其原因主要有3个:①新生儿巨结肠确诊困难,一般认为新生儿巨结肠的形态学改变,生后2周才形成,有的需要3~4周甚至几个月。尽管开展了灌肠后24~48h,动态观察钡剂贮留或排泄的功能性改变,但梗阻症状很重时,钡剂灌肠后必须洗肠或手术,...[详细]
 2.直肠肛管测压 是诊断先天性巨结肠的有效方法,具有经济、简便、快速而安全,以及无损伤性,可反复检测等优点。关于测压诊断先天性巨结肠的准确率,文献报道不一(76%~100%)。研究证明,正常儿直肠内气囊注入2~3ml气体后,1~3s内肛管压力迅速下降(称正常反射),而先天性巨结肠病儿,向直肠内气囊注入很多气体,肛管压力都不变(称阴性反射),即无直肠肛管反射或无正常反射,有的先天性巨结肠病儿,肛管压力不但不下降,反而上升(称异常反 射)(图2)。阴性反射和异常反射统称为病理反射,经检测156例慢性便秘病儿,直肠肛管测压的准确率为93.33%,其中误诊率为2.88%(104例病理反射者,3例除外先天性巨结肠:2例为新生儿,6个月后复查正常;另1例为胎粪性腹膜炎),漏诊率为7.69%(52例有正常反射者,4例为先天性巨结肠病儿)。为提高测压诊断的准确性,必须注意检测方法和判断标准。
2.直肠肛管测压 是诊断先天性巨结肠的有效方法,具有经济、简便、快速而安全,以及无损伤性,可反复检测等优点。关于测压诊断先天性巨结肠的准确率,文献报道不一(76%~100%)。研究证明,正常儿直肠内气囊注入2~3ml气体后,1~3s内肛管压力迅速下降(称正常反射),而先天性巨结肠病儿,向直肠内气囊注入很多气体,肛管压力都不变(称阴性反射),即无直肠肛管反射或无正常反射,有的先天性巨结肠病儿,肛管压力不但不下降,反而上升(称异常反 射)(图2)。阴性反射和异常反射统称为病理反射,经检测156例慢性便秘病儿,直肠肛管测压的准确率为93.33%,其中误诊率为2.88%(104例病理反射者,3例除外先天性巨结肠:2例为新生儿,6个月后复查正常;另1例为胎粪性腹膜炎),漏诊率为7.69%(52例有正常反射者,4例为先天性巨结肠病儿)。为提高测压诊断的准确性,必须注意检测方法和判断标准。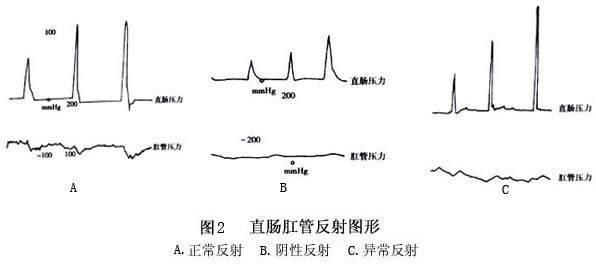 直肠肛管测压诊断新生儿巨结肠应当慎重。有作者动态检测50例正常新生儿,仅13例于生后第1天出现正常反射,48例(96%)在生后1周内出现正常反射,另2例因出院未能连续检测,分别在生后100天和8个月测检时出现正常反射。理论上应该说,新生儿生后自动排便,标志有直肠肛管反射,但经测压观察,我们认为这种刚刚形成或建立的反射,并未成熟也不稳定,故测检中不易显示或捕捉。 目前一致认为,诊断和鉴别超短段先天性巨结肠与特发性巨结肠的最可靠方法,是直肠肛管测压检查。
直肠肛管测压诊断新生儿巨结肠应当慎重。有作者动态检测50例正常新生儿,仅13例于生后第1天出现正常反射,48例(96%)在生后1周内出现正常反射,另2例因出院未能连续检测,分别在生后100天和8个月测检时出现正常反射。理论上应该说,新生儿生后自动排便,标志有直肠肛管反射,但经测压观察,我们认为这种刚刚形成或建立的反射,并未成熟也不稳定,故测检中不易显示或捕捉。 目前一致认为,诊断和鉴别超短段先天性巨结肠与特发性巨结肠的最可靠方法,是直肠肛管测压检查。诊断
诊断先天性巨结肠,主要根据临床表现,确诊则需要X线钡剂灌肠、直肠肛管测压、直肠活检、组织化学等客观检查方法。 新生儿巨结肠的症状需要与胎粪性便秘、肠闭锁或狭窄、肛门直肠畸形等病鉴别。用小指肛诊后,巨结肠病儿能大量排出胎便及气体,随之症状缓解。因此,对新生儿肠梗阻,常规做肛诊检查,不仅有助于诊断和治疗新生儿巨结肠,而且可避免或减少错误诊治。 6个月以上病儿,有慢性便秘等症状、又有新生儿巨结肠史,诊断先天性巨结肠较容易,否则须除外肛门直肠畸形等病所致续发性巨结肠,以及克汀病、饮食性便秘、特发性巨结肠等症。肛诊时,短段型先天性巨结肠的肛门紧缩,直肠明显扩张积便或积气;普通型先天性...[详细]
治疗
1.内科治疗?对轻型的先天性巨结肠患儿或有全身感染症状,手术无法耐受者可用非手术疗法维持营养和发育。用缓泻剂或定时用生理盐水灌肠,以避免粪便淤积。 2.结肠造瘘术?当患儿发生急性肠梗阻,或有肠穿孔、腹膜炎趋向,或伴有小肠结肠炎,或是全结肠无神经节症,应行结肠造瘘术。造口部位应尽量选择靠近扩张肠管作单口造瘘。 3.手术治疗?新生儿期患儿应尽量保守治疗,待1岁左右再做根治手术。成人期患者症状加重,经保守治疗无效者亦应行根治术。根治手术要求切除距肛门齿状线1~2cm处以上的狭窄直肠及狭窄段上5cm以上的扩张结肠。常用的术式有下面三种: (1)拖出型直肠、乙状结肠切除术(...[详细]
预后
新生儿HD诊断治疗均十分困难,多数文献报道,采用常规洗肠等保守疗法,半年内病死率为50%~70%,1年达70%~90%。肠炎发生率为20%~30%左右,肠穿孔约为3.4%~6.4%。国内余氏亦报道新生儿保守治疗及肠造瘘术后总死亡率仍高达40%。新生儿根治手术死亡率为3.1%~12%,近年来也有少数病例报道根治术未发生死亡者。因此对新生儿的HD诊治应特别慎重,根据患儿一般情况及病变肠管的长度、医院设备及条件,可分别选择中西医结合非手术疗法,经肛门路手术及根治手术。 虽然婴幼儿HD随着年龄的增长,其手术危险性逐渐降低。但是根据国外大宗病例报道,根治术后并发症仍然较多。术后伤口感染约占10...[详细]
预防
目前没有相关内容描述。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号



